Received: Wed 06, Dec 2023
Accepted: Mon 22, Jan 2024
Abstract
Myopathies present a wide range of clinical symptoms that affect the skeletal muscles, including weakness, fatigue, and pain. While acquired myopathies receive significant attention due to the availability of treatment options, it is important to note that some inherited myopathies can also be effectively managed. These myopathies can be classified based on their underlying causes, such as infectious agents, autoimmune disorders leading to muscle inflammation, granulomatous inflammation, metabolic abnormalities within the muscle cells, skeletal muscle channel dysfunctions, prolonged ICU stay, and inherited conditions such as Duchenne muscular dystrophy. In this review, we initially present a clinical approach to neuromuscular diseases and subsequently place specific emphasis on myopathies, particularly to those that have treatment options available.
Keywords
Myopathy, treatable, neuromuscular disorders, nmd, approach, muscle weakness
1. Introduction
Neuromuscular disorders (NMD), characterized by muscle weakness, are classified within the lower motor neuron syndrome (LMN) [1]. These disorders can be categorized based on the specific level affected, including the motor neuron (neuronopathies), nerve root/plexus (radiculoplexus neuropathies), individual peripheral nerves (focal/multi-focal/diffuse polyneuropathies), neuromuscular junction (NMJ disorders), and muscle (myopathies). Myopathies encompass a broad spectrum of disorders that primarily affect the skeletal muscles, leading to weakness, fatigue and pain. These conditions can be either acquired or inherited, with varying etiologies and clinical presentations. Myopathies can arise from diverse factors, including infectious agents, autoimmune processes, metabolic abnormalities, genetic mutations and other underlying systemic diseases. Understanding the underlying mechanisms and classifying myopathies based on their etiology is crucial for accurate diagnosis and appropriate management.
In this review, we will provide an overview of myopathies, highlighting their diverse clinical manifestations and categorization by etiology. We will explore the different types of myopathies, ranging from infectious and autoimmune myopathies to metabolic disorders, channelopathies and genetic conditions like Duchenne muscular dystrophy. By delving into the various aspects of myopathies, we aim to shed light on the complexities of these disorders and emphasize the importance of targeted approaches for their diagnosis, treatment, and ongoing management. Myopathies present a significant burden on individuals affected by these conditions, as well as on healthcare systems worldwide. Advances in research and medical interventions have enabled improved understanding, diagnosis, and therapeutic options for some myopathies. However, challenges remain in identifying and effectively treating certain types of myopathies, particularly those with genetic origins. Therefore, ongoing research and multidisciplinary efforts are essential to further unravel the complexities of myopathies and develop innovative strategies to enhance patient care and quality of life.
In the following sections, we will explore the different categories of myopathies, discuss their etiologies, clinical features, and available treatment options. We aim to provide a comprehensive overview that will serve as a valuable resource for healthcare professionals, researchers and individuals affected by myopathy.
2. Diagnostic Clues for Neuromuscular Diseases
2.1. Clinical History
When taking a history in the context of neurologic disciplines, it is important to consider specific aspects. Paying close attention to the onset of symptoms is crucial, such as whether they occurred acutely, or if the limb weakness is symmetrical. Additionally, tracking the appearance of new symptoms over time, and identifying any precipitating factors or activities that have been limited are important. It is essential to assess whether the course of the condition is chronic and progressive or if it exhibits a diurnal pattern and is related to fatigue and exercise intolerance. Gathering information about past or co-morbid illnesses, along with a comprehensive medication list, is necessary. Understanding the patient’s baseline function is also valuable. A caveat is that patients may have been suffering from NMD for a long time that their disabilities lead to negative or difficult attitudes toward the disease. Obtaining a 3-generation family history of NMD through a genogram is ideal for determining autosomal dominance, consanguinity in reference to autosomal recessive inheritance, as well as identifying the x-linked disorders. However, challenges may arise in determining family history, including situations where family members have passed away at an early age, families with only one child, adoption scenarios, variations within the same family, and milder forms of the disease that may manifest.
2.2. General Physical Examination
The presence of cardiomyopathies and arrhythmias often point to inherited myopathies such as dystrophinopathies, limb-girdle muscular dystrophies (LGMD), myotonic dystrophies and Andersen–Tawil syndrome. Metabolic glycogen (GSD) and lipid storage myopathies may present with hepatosplenomegaly. Inherited neuropathies may present with skeletal deformities like pes cavus and kyphoscoliosis. Deafness, retinitis pigmentosa and short stature allude to mitochondriopathies. Joint laxity with or without contracture indicate collagenopathies like Ullrich congenital muscular dystrophy. Skin rashes and ‘mechanic’s hands’ point to immune-mediated disorders, like dermatomyositis, while skin pigmentation points to POEMS (polyneuropathy, organomegaly, endocrinopathy/edema, M-proteinemia and skin changes) [2], adrenal failure or B12 deficiency. Palm and sole hyperkeratosis are noted in toxic neuropathies, (e.g., arsenic toxicity, and their nails show Mee’s lines). Autonomic neuropathy is considered in cases with hair loss in distal limbs, as are cold feet and hands.
2.3. Neurologic Examination
Clinical evaluation can provide valuable clues in assessing muscle weakness. Some key observations include asking the patient to stand erect and walk normally to observe for ataxia such as sensory ataxia in Romberg’s test. Assessing the ability to walk on heels and toes helps evaluate distal-dominant weakness, while the patient’s ability to rise from a chair or ground, and lift arms overhead provide insight into proximal-dominant weakness. Weakness of the knees results in hyperextension and the genu recurvatum and high steppage gait point to foot drop, whether symmetric or asymmetric. Exaggerated lordosis and protuberant abdomen can point to truncal muscle weakness, while a waddling gait may indicate gluteal weakness. Axial muscle involvement is indicated by the presence of an abnormal posture, rigid spine or contractures, a dropped head, bent spine (camptocormia) and atrophy in the paraspinal muscles [3].
Scapular winging from atrophy can be elicited by asking patient to outstretch both upper limbs and to rest both palms on the wall. Neck muscle weakness, especially in flexion, can be a useful test, along with manual muscle examination focusing on specific proximal and distal joint movers. Facial weakness, characterized by drooping or lagging of facial expression and mimic movements, jaw bite and deviation, tongue at rest (for atrophy and fasciculations) and protrusion (for deviation). Specific eye movement impairment (ptosis/weak eye movements [EOM]), including eye closure observed in sleep) are helpful clues. Muscle hypertrophy which is best seen in gastrocnemius, hip, shoulder and elbow movers, for preferential muscle group/myotomal atrophy, and for abnormal movements like fasciculations (brief scattered twitches at irregular intervals; usually seen in chest, shoulder, upper back, proximal limbs and tongue), myokymia (muscle quivering, undulating and slower contraction of muscle strips), and rippling muscles (provoked by mechanical stimuli and stretch). Deep tendon reflexes (DTRs), assessed with a reflex hammer are important for evaluating reflex symmetry and ruling out upper motor neuron signs. In addition to grip myotonia, a reflex hammer is also used to elicit percussion myotonia by tapping the thenar eminence and finding delayed relaxation. Basic sensory examination will assist in eliminating neuropathic disorders.
Table 1 presents the summary of the neurologic examination for NMDs. Table 2 presents an algorithm that can serve as a guide in the localization of the lesion when confronted with a patient with muscle weakness. The foremost clinical approach is to ensure that weakness is not attributable to central or associated long tracts (i.e., with UMN signs) like spasticity; or with bradykinesia and rigidity (i.e., in Parkinsonism); or with cerebellar ataxia. If the clinical manifestations cannot be explained by the aforementioned levels, a functional neurological disorder (FND) should be considered.
TABLE 1: Summary of neurologic examination for NMDs.
|
-Assess standing
posture and walking for ataxia |
|
-Evaluate
distal-dominant weakness by walking on heels and toes |
|
-Test ability to rise
from a chair or ground, and lift arms overhead for proximal-dominant weakness |
|
-Look for
hyperextension and genu recuvatum indicating weakness of the knee |
|
-Check for high
steppage gait suggesting foot drop |
|
-Examine for
exaggerated lordosis, protuberant abdomen, and waddling gait |
|
-Look for abnormal
posture, rigid spine, contractures, dropped head, paraspinal muscle atrophy
indicating axial muscle involvement |
|
-Assess scapular
winging by asking the patient to outstretch both upper limbs |
|
-Test neck muscle
weakness, facial weakness, and eye movement impairments |
|
-Look for muscle
hypertrophy, myotonic movements, fasciculations, myokymia and rippling
muscles |
|
-Assess deep tendon
reflexes and look for grip myotonia and percussion myotonia |
|
-Perform basic
sensory examination to rule out neuropathic disorders |
TABLE 2: Flow chart of approach to muscle weakness in NMDs
|
Muscle weakness? -YES (but NO UMN signs,
Parkinsonism and Ataxia) -YES (but NO local
bone, joint and tendon pains) -YES (but NOT feigned
as in Functional Neurological Disorder)
>Assess STEP-1: History, physical/systemic and
neurologic examinations
I. Is there SENSORY involvement? A. YES, With sensory involvement
(Deficits/Neuropathic pain) [80] 1. Neuronopathy: Herpes
Zoster, B6 toxicity, Paraneoplastic syndrome 2. Neuropathy: Asymmetric Peripheral (Radiculopathy,
Plexopathy, neuropathy [Hansen’s, Vasculitic, compressive/trauma, diabetic,
Bell’s palsy, Sarcoidosis); vs Symmetric
Peripheral (Metabolic [diabetic, uremic], Infectious [Lyme’s, HIV, COVID19],
Post-infectious/vaccinal/Autoimmune [GBS/CIDP, COVID19 vaccine [81]],
Paraproteinemic [2] , Paraneoplastic [82], Toxic [drugs, heavy metal] and
Hereditary [CMT, Amyloidosis, Fabry’s])
B. NO sensory involvement 1. Neuronopathy: Symmetric (SMAs); vs Asymmetric/Segmental (Poliomyelitis,
Hirayama Disease; and MND [ALS if w/ UMNs]) 2. Neuropathy: MMN [83] 3. NMJ: MG, CMS, LEMS,
BTX (Botulism) 4. Myopathy: Proximal-dominant (Muscular
dystrophies, Limb-girdle Syndromes (LGMD), Congenital myopathies, Muscle
Channelopathies, Inflammatory [infectious, autoimmune], Toxic [drugs], and
Metabolic myopathies [84] [Hypokalemia, Thyroid, Glycogen and Lipid
Storage]); vs Distal-dominant
(Distal Myopathies, Myotonic dystrophy, IBM)
>Assess STEP-2: Pattern Recognition
II. Is there FACIAL Muscle Weakness?-YES 1. Neuronopathy:
Kennedy’s Disease [83] 2. Neuropathy:
Asymmetric (Bell’s Palsy, Diabetic [85], Sarcoidosis [86], Hemifacial spasm);
vs Symmetric (GBS, CIDP) 3. NMJ: MG, CMS 4. Myopathy: FSHD,
Myotonic dystrophy, Central Core disease, Mitochondrial disease
III. Is there OCULAR and/or PHARYNGEAL Muscle
Weakness?-YES 1. Neuronopathy: ALS,
Kennedy’s Disease 2. Neuropathy: GBS
(Miller-Fisher and Pharyngeal-Cervical-Brachial variants), CIDP, Diphtheric
neuropathy 3. NMJ Dis: MG, CMS,
LEMS, BTX 4. Myopathy:
Inflammatory myopathy (autoimmune; Pharyngeal), Myotonic dystrophy (Ocular),
Centronuclear myopathy (Ocular), OPMD, OPDM, Thyroid (ocular) and
Mitochondrial disease
IV. Is there PARASPINAL
Muscle Weakness? -YES 1.Neuronopathy: ALS 2.Neuropathy: Diabetic
cervical/lumbosacral radiculoplexus neuropathy [87] 3.NMJ: MG 4.Myopathy:
Calpainopathy, Dysferlinopathy, FSHD, LGMD, MD 1, IBM [3]
>Assess STEP-3:Is Weakness related to Fatigue and
Exercise
V. Is Weakness EPISODIC and FATIGUE-RELATED?-YES 1. NMJ Dis: MG, CMS,
LEMS, BTX 2. Myopathy:
Hypokalemic Periodic Paralysis, Thyrotoxic Periodic Paralysis, Andersen-Tawil
syndrome, Glycogen storage diseases
VI. Is there MYALGIA and/or Exercise-induced
CRAMPS?-YES 1. Neuronopathy: ALS,
Kennedy’s Disease, Post-polio syndrome 2. Neuropathy:
Radiculopathy, Diabetic and Uremic neuropathies, CMT, Isaac’s disease 3. NMJ: BTX 4. Myopathy:
Rhabdomyolysis, Inflammatory myopathy (autoimmune), Myotonic dystrophy,
Muscle Channelopathies, Toxic (drugs), Metabolic (Thyroid, Glycogen and Lipid
Storages dis) and Mitochondrial diseases
VII. Is there Clinical MYOTONIA
(Grip/Percussion)?-YES 1. Neuropathy: Isaac’s
syndrome (grip myotonia) 2. Myopathy: Myotonic
Dystrophy and Muscle Channelopathies (Grip & Percussion) ------------------ |
ALS:
Amyotrophic Lateral Sclerosis; BTX: Botulism; CMS: Congenital Myasthenic
Syndrome; CMT: Charcot-Marie tooth disease; FSHD: Facioscapulohumeral Myotonic
Dystrophy; GBS/CIDP: Guillain Barre Syndrome/ Chronic Inflammatory
Demyelinating Polyneuropathy; IBM: Inclusion Body Myositis; LEMS: Lambert-Eaton
Myasthenic Syndrome; LGMD: Limb Girdle Muscular Dystrophy; MD: Myotonic Dystrophy;
MND: Motor Neuron Disease; NMJ: Neuromuscular Junction; MG: Myasthenia Gravis;
OPMD: Oculopharyngeal Muscular Dystrophy; OPDM: Oculopharyngodistal Myopathy;
SMA: Spinal Muscular Atrophy; UMN: Upper Motor Neuron
3. Classification of Myopathies
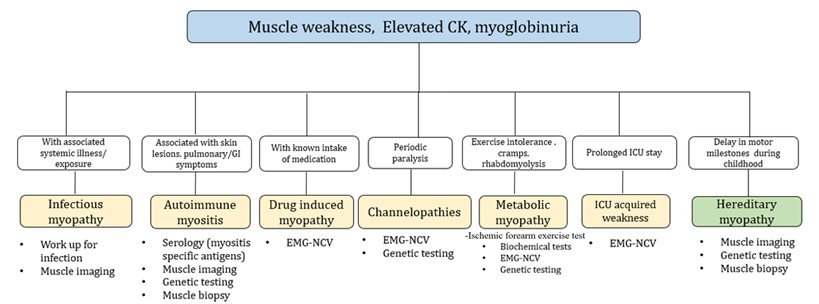
Myopathies encompass a diverse range of conditions characterized by abnormalities in the structure or function of skeletal muscle and can be classified as acquired or hereditary. Table 1 provides a comprehensive guide to distinguishing myopathy from other neuromuscular disorders (NMDs). While acquired and acute myopathies are prioritized due to the availability of treatment options, it is important to recognize that certain inherited myopathies can also be effectively treated. These treatable myopathies can be classified based on their etiology, including infectious myopathies, granulomatous myopathies, autoimmune myopathies, metabolic myopathies, skeletal channel myopathies, and Duchenne muscular dystrophy (DMD). The initial laboratory assessment of patients suspected of having myopathy often includes measuring the creatine kinase (CK) level. However, it is important to note that a normal CK level does not exclude the presence of myopathy, particularly in cases of slowly progressive conditions, significant muscle atrophy, or individuals receiving corticosteroid therapy [4, 5].
Other muscle-associated enzymes, although non-specific, can be helpful in patients with a suspected myopathy, including aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, and aldolase [5, 6]. Although a myopathic electrodiagnosis may be established by showing electromyographic (EMG) brief, low amplitude polyphasic potentials, early recruitment and whether or not coupled with irritative potentials, this diagnostic test specificity is low. Nowadays, muscle imaging techniques such as ultrasound and magnetic resonance imaging (MRI) play a valuable role in the evaluation and differentiation of myopathies [7, 8]. An algorithmic classification of treatable myopathies and recommended diagnostic work up is made in (Figure 1). Not to be missed treatable myopathies are periodic paralysis, rhabdomyolysis and those that occur in the intensive care.
3.1 Acquired Myopathies
3.1.1. Infectious Myositis
Myositis refers to the inflammation of muscle tissue, and although muscles are generally resistant to infection, there is a wide range of organisms that can cause this condition, including viruses, parasites, bacteria and fungi [9]. This present review will only delve into the most common causative organisms, certainly not encompassing several other causes of infectious myositis found to date. The clinical course of myositis can vary, presenting as acute, subacute or chronic, and may be accompanied by systemic manifestations like fever, rash and thrombosis. In bacterial myositis, hematogenous spread and direct invasion like trauma, surgery, exposure to contaminated soil play important roles in the development of the condition. Immunosuppression also increases susceptibility to certain bacterial and fungal organisms. Initially, these infections may affect specific muscles, then may spread to other areas, if the microbial load is overwhelming. In the case of viral myositis, generalized muscle weakness and fatigue are more commonly observed, and since the COVID-19 pandemic, there have been reports of acute and post-infectious muscle involvement [10]. The diagnosis of myositis relies not only on the clinical presentation and possibly imaging studies but also on confirmation through serologic tests and cultures, as identifying the underlying pathogen is crucial for appropriate treatment selection.
3.1.1.1. Viruses
Viruses cause infectious myositis, through mechanisms such as direct cytopathic effect, immune complex formation, and immune dysregulation [11]. Muscle damage primarily occurs due to direct myotoxic effects. Due to their trophic property towards immature muscle cells, viral myositis is more commonly observed in children [12]. Symptoms include myalgia, symmetric weakness, and rhabdomyolysis, often preceding a gastrointestinal or respiratory infection influenza A, influenza B, coxsackievirus, Epstein-Barr virus are common culprits. Notably, since the onset of COVID-19 pandemic, reports have emerged regarding myositis associated with SARS-Cov-2 infection. Factors contributing to weakness and fatigue in these cases include prolonged hospitalization, malnutrition, disuse and hypoxemia [9, 13]. Perhaps a direct invasion of muscle tissue by the virus, coupled with adaptive immune response mediated by the angiotensin-converting enzyme ACE2 receptor, triggers myositis in COVID-19 patients [13, 14].
Recent literature indicates that rhabdomyolysis has been observed in both adult and pediatric individuals infected with COVID-19 [15]. This pathogenesis is linked to myotoxic cytokines including C-X-C motif chemokine 10, interferon gamma, interleukin 1 beta (IL-1β), IL-6, IL-8, IL-17, and tumor necrosis factor alpha [13]. Cases of myositis following COVID-19 vaccination have also been reported and associated with anti-Jo1 antibody [16]. Furthermore, muscle weakness and fatigue persist in 50-60% of patients even six months after the initial infection [14]. Elevated CK levels in conjunction with muscle weakness are typically present. Notably, patients with severe COVID-19 infection admitted to the intensive care unit exhibited a 30% reduction in the cross-sectional area of the rectus femoris, with a decreased thickness of the quadriceps muscle after 10 days [17]. While antiviral therapies such as ritonavir, nirmatrelvir, remdisivir, and molnupiravir, as well as vaccines, are available to combat and prevent COVID-19, when it comes to managing COVID-19 myopathy, prioritizing physical therapy and early rehabilitation remain essential [15].
Endemic chronic retroviral infections such as human T-lymphotrophic virus, type 1 (HTLV-1), have been implicated in myositis. Studies of muscle biopsy specimens, have revealed HTLV-1 in CD4+ cells rather than macrophages, indicating that the majority of HTLV-1-containing CD4+ cells are lymphocytes rather than macrophages [18]. Treatment approaches for HTLV-1 infections, including myositis, lack systematic studies and consists of a range of medications such as steroids, interferon, danazol, high-dose vitamin C, azathioprine and antivirals commonly used in human immunodeficiency virus (HIV) infection (e.g. lamivudine and zidovudine, the latter of which can induce mitochondrial toxicity leading to myopathy). Myopathy, infiltrative lesions and rhabdomyolysis can also manifest in individuals with HIV infection. Prompt recognition and initiation of treatment yield a favorable prognosis for HIV-associated myositis.
3.1.1.2. Bacterial Infections
Bacterial infections of the muscle typically occur through direct invasion from trauma and hematogenous spread. Staphylococcus aureus is the main cause of pyomyositis [11], characterized by localized pain, tenderness of the affected muscle, fever and potential abscess formation. If untreated, pyomyositis can lead to complications such as osteomyelitis, endocarditis and sepsis. Notably, typical findings in soft tissue infections like lymphadenitis and focal erythema, are absent in pyomyositis. Diffuse muscle involvement occurs in 10-20% of cases [11]. Some strains of Staphylococcus aureus produce exotoxins that cause toxic shock syndrome, believed to be due to T-cell activation and cytokine release. Infections caused by Group A B-hemolytic streptococcus, though less common are more severe, leading to necrotizing fasciitis and streptococcal toxic shock syndrome. These infections are often opportunistic and more prevalent among individuals with diabetes and those with compromised immune systems. Polymicrobial infections are also common in cases of vascular insufficiency and penetrating wounds. Gram stain and aerobic/anaerobic cultures should be performed immediately to guide antibiotic therapy. Penicillins, cephalosporins, and vancomycin with the addition of gentamicin and clindamycin should be initiated even without culture results. In cases where necessary, surgical drainage of pus should be performed alongside intravenous antibiotic treatment.
3.1.1.3. Parasitic Myositis
Parasitic myositis [19] should be suspected in patients presenting with muscle aches, a history of travel to endemic areas, and ingestion of undercooked meat. Common causes include Taenia solium, Trichinella species and Toxoplasma gondii. Symptoms include weakness, swelling and myalgia, along with elevated CK, eosinophilia, erythrocyte sedimentation rate and C-reactive protein. Diagnosis can be confirmed through muscle biopsy and serologic tests. Taenia solium, or pork tapeworm, is acquired from ingesting Taenia solium eggs from undercooked pork and can cause not only cause gastroenteritis but also the dissemination of larvae via the bloodstream, leading to cysticercosis. Neurocysticercosis can occur when the larvae reach the central nervous system and is one of the most common causes of adult-onset seizures [20]. In one population study in rural Ecuador by Brutto et al., [21], individuals with neurocysticercosis were six times more prone to develop adult-onset epilepsy than those without the disease [21]. Trichinella larvae cause chronic infections in the muscle. Trichinosis also occurs with ingestion of encysted larvae from undercooked boar, pork or dog meat, forming a capsule inside the muscle, leading to repeated regeneration, degeneration and necrosis. Serologic tests are done with anti-trichinella IgG antibodies. Protozoal toxoplasmosis from Toxoplasma gondii cysts can be acquired from consuming food contaminated with oocysts from cat feces and may infect the muscles. Immunocompromised individuals with low CD4 counts are particularly susceptible to Toxoplasma gondii infection. Treatment of the muscle involvement include specific medications such as praziquantel, albendazole, and mebendazole for helminthic infections and pyrimethamine, sulfadiazine, atovaquone for protozoal infections. In some cases, surgical removal of the lesion may be necessary.
3.1.1.4. Fungal Myositis
Fungal myositis is most commonly seen in with immunocompromised patients [11]. It typically affects a single muscle or group of muscles due to abscess formation. The clinical presentation resembles bacterial myopathies. Candida is the most frequent cause of fungal myositis [9]. Risk factors include use of broad-spectrum antibiotics, immunosuppressive drugs, and severe neutropenia, leading to diffuse muscle tenderness accompanied by rash and fever. Histologic examination reveals budding yeast and pseudohyphae. Cryptococcus myositis is uncommon and is found in diabetic and immunocompromised patients presenting with a disseminated cryptococcal disease. Histoplasmosis rarely causes myositis after inhalation of dimorphic fungi post-dissemination to the lungs. Therapeutic anti-fungal regimens include amphotericin B and triazoles (fluconazole, itraconazole, voriconazole, ravuconazole) administered orally or parenterally. Echinocandins may be used such as caspofungin, micafungin, as are the antimetabolites like flucytosine.
3.1.1.5. Granulomatous Myositis
Granulomatous myositis is a rare disease characterized by presence of non-caseating granulomatous inflammation in the skeletal muscle. It can occur spontaneously or be associated with inflammatory conditions such as sarcoidosis, primarily affecting females in their fifth decade. Common symptoms include dysphagia and bilateral and symmetric muscular weakness, often proximal in nature [22]. In this review we discuss sarcoid myopathy which is the most common cause of granulomatous myositis.
Sarcoidosis is a multisystem disorder characterized by noncaseating granulomas in various organs. Musculoskeletal involvement is less common, and when present, it typically accompanies other organ involvement. The pathogenesis of sarcoidosis is not well understood, but it is characterized by the accumulation of T lymphocytes, mononuclear phagocytes, and noncaseating granulomas [23]. Chronic myopathy, the most common form of sarcoid myopathy, presents with progressive symmetric proximal muscle weakness. The other forms include acute sarcoid myositis, characterized by diffuse muscle swelling and nodular myopathy which present with single or multiple, bilateral, tender nodules, usually found on the lower limbs. Nodules vary in size and are palpable, painful and is not associated with muscle weakness. Diagnosis includes elevated total CK, myopathic EMG, and ultrasound and MRI findings, and muscle biopsy showing granulomatous infiltration. Treatment of sarcoid myopathy involves the use of glucocorticoids (prednisone), hydroxychloroquine, and immunosuppressive agents (methotrexate, azathioprine or tumor necrosis factor inhibitors). Glucocorticoids are tapered over one year, and immunosuppressive medications are continued for two to three years. Prognosis depends on the early management of the myopathies; however, these are not expected to be fatal. A chronic myopathy may lead to long term functional decline due to muscle atrophy [23].
3.1.2. Autoimmune Myositis
Autoimmune myositis represents a large and diverse group of treatable myopathies. The recent classification of these conditions incorporates clinical and pathological, and immunologic markers. Currently, autoimmune myositis is categorized into several subtypes, including dermatomyositis, inclusion body myositis (IBM), immune-mediated necrotizing myopathy (IMNM), antisynthetase syndrome (ASS) and overlap myositis [24-26]. The clinical presentation often involves proximal muscle weakness, such as difficulty climbing stairs or raising both arms, accompanied by dermatologic and pulmonary manifestations. However, in IBM, asymmetric weakness of long finger flexors, as well as anterior thigh muscles is typical. While muscle biopsy provides valuable pathologic information, the testing for autoantibodies (i.e., myositis-specific and myositis-associated antibodies) has become increasingly important in differentiating between these diseases (Table 3). Genetic studies have been conducted to identify risk factors and dysregulated gene expression in autoimmune myositis, aiming to understand the disease's underlying mechanisms. Genome-wide association studies (GWAS) have confirmed the human leukocyte (HLA) region as strongly associated with autoimmune myositis, with different associations observed among clinical subgroups [27]. Rare genetic variation, such as mitochondrial DNA variation and somatic mutations, have been found to contribute to disease susceptibility in specific autoimmune myositis subtypes and these mutations are thought to contribute to the pathogenesis of the disease [27].
TABLE 3: Autoantibodies in autoimmune myositis.
|
Dermatomyositis |
|
|
Anti-nuclear matrix protein (NXP 2) |
Subcutaneous calcinosis and typical skin lesions of dermatomyositis,
common in children |
|
Anti-transcription intermediary factor 1 Y (TIF1-Y) |
Increased risk of malignancy (Breast, lung, gastric, colorectal,
thymus, ovary) and typical skin lesions of dermatomyositis |
|
Anti-melanoma differentiation associated gene 5 (MDA 5) |
Atypical skin lesions, rapidly progressive interstitial lung disease,
CADM |
|
Anti–complex nucleosome remodeling histone deacetylase (Mi-2) |
Typical skin lesion of dermatomyositis. Clinical course is benign |
|
Anti-small ubiquitin- like modified activating enzyme (SAE) |
Associated with typical skin lesions of dermatomyositis with mild
myopathy |
|
Immune mediated necrotizing myopathy |
|
|
Anti-HMGCR |
Associated with statin intake but may be seen in statin-naïve patients
especially in Asian countries, better response to immunosuppression |
|
Anti-SRP
|
Associated with more severe muscle weakness, poor treatment response |
|
Inclusion body myositis |
|
|
Anti cystosolic 5’ nucleotidase 1A |
Weakness in finger flexors and knee extensors |
|
Antisynthetase syndrome |
|
|
Anti-Jo 1 (Antihistidyl) |
Associated with fever, ILD, arthritis, Raynaud’s phenomenon and
mechanic’s hand |
|
Anti-PL 7 (Antithreonyl) |
|
|
Anti-PL 12 (Antialanyl) |
|
|
Anti-OJ (Antiisoleucyl) |
|
|
Anti-EJ (Antiglycyl) |
|
|
Anti-KS (Antiasparaginyl) |
|
|
Anti-Zo (phenylalanyl) |
|
|
Anti-Ha (Antityrosyl). |
|
Typical skin lesion of
dermatomyositis: Periorbital edema, heliotrope rash, Gottron’s sign, V-sign,
shawl sign
Atypical rash:
mechanic’s hands, palmar papules, ulceration of the fingers
CADM: clinically
amyopathic dermatomyositis
ILD: Interstitial lung disease
*15-30%
Prevalence in myositis [28].
3.1.2.1. Dermatomyositis (DM)
Dermatomyositis (DM) can occur in both children or adults and is characterized by a gradual onset of proximal muscle weakness and distinct cutaneous manifestations. These include periorbital edema, heliotrope rash, Gottron’s sign, V-sign and Shawl sign. The pathogenesis of the disease involves a range of intrinsic and extrinsic factors, encompassing genetic, environmental, as well as immune and non-immune-mediated factors [29]. Genetic predisposition plays a significant role in the development of DM. Genotyping studies have shown associations between variations in the major histocompatibility complex (MHC) and DM susceptibility [29, 30]. Specifically, certain HLA alleles have been correlated with the production of autoantibodies in both adults and children. Epigenetic modifications include histone modification, DNA methylation and microRNA activity [30]. There is also genetic overlap with autoimmune disorders [27]. In the study made by Miller et al., they evaluated 269 SNPs associated with rheumatoid arthritis, systemic lupus erythematosus (SLE), type 1 diabetes mellitus, Crohn's disease, thyroid disease, gluten-sensitive enteropathy, or multiple sclerosis. Three genes (PLCL1, BLK, and CCL21) showed significant associations with DM, despite not being previously associated with the condition [30].
Recent clinicopathologic subtypes include clinically amyopathic dermatomyositis (CADM), where classic DM skin lesions are present without muscle involvement, dermatomyositis sine dermatitis, which exhibits clinical and pathologic evidence of DM but lacks skin lesions and juvenile dermatomyositis (JDM) in individuals under 18 years of age [24]. Total CK levels are commonly elevated, and can increase up to 50 times in active disease [31]. The myositis-specific antibodies (MSA) such as anti-Mi2, anti-SAE, anti-MDA5, anti-TIF1, and anti-NXP2 are specific for DM [32]. These various circulating antibodies, each carrying its own diagnostic implications (Table 3). For instance, anti-Mi2 antibodies are linked to classic DM, while anti-TIF1 antibodies are strongly associated with underlying malignancy. Anti-MDA5 antibodies indicate an increased risk of rapidly progressive interstitial lung disease (RP-ILD). Anti-NXP-2 antibodies are associated with calcinosis and severe myopathy, especially in juvenile DM. Anti-SAE antibodies are found in a subset of DM patients with severe cutaneous involvement and minimal myopathy [32]. The key histopathological feature of DM observed on muscle biopsy is perifascicular atrophy according to Dalakas et al. [31].
Although highly specific, its sensitivity remains around 25-50% [33]. The cellular infiltrate consists primarily of perivascular macrophages, B cells, CD4 cells, and plasmacytoid dendritic cells. The activation and accumulation of complement proteins lead to the destruction of endomysial capillaries and in muscle ischemia which can be observed with membrane attack complex (MAC) deposition on the sarcolemma. Sarcoplasmic expression of myxovirus resistance protein A (MxA) is specifically observed in all types of DM, indicating the crucial involvement of type I interferons in its pathogenesis [34]. MxA serves as a sensitive diagnostic marker, with a sensitivity and specificity of 71% and 98% respectively, surpassing the significance of perifascicular atrophy [36]. In the study made Uhura et al. [35], sarcoplasmic MxA expression is seen in both juvenile and adult DM, suggesting its value as a diagnostic marker regardless of age. MRI of the muscle often reveal fascial edema [24, 36]. The prognosis of patients with DM is generally good, with a reported 70%-93% survival rate at 5 years. However, the presence of poor prognostic features such as malignancy or lung involvement can decrease this rate [36].
3.1.2.2. Immune-Mediated Necrotizing Myopathy (IMNM)
Immune-mediated necrotizing myopathy (IMNM) is a distinct subtype of autoimmune myositis characterized by severe proximal weakness, myofiber necrosis with minimal inflammatory cell infiltrate on muscle biopsy, and limited extra-muscular involvement. To date, there are two distinct autoantibodies found in patients with IMNM. One targets 3-hydroxy-3-methylglutaryl-CoAreductase (HMGCR), while the other targets signal-recognition peptide (SRP) [37]. Anti-HMGCR is associated with statin exposure [38, 39]. Studies conducted on predominantly caucasian populations have shown a higher occurrence of statin exposure in anti-HMGCR+ immune-mediated necrotizing myopathy (IMNM) compared to Asian cohorts [40]. While IMNM is commonly observed in individuals aged 30 to 70 years, it can also affect pediatric patients [41]. The pathogenesis of IMNM is multifactorial, involving immune responses triggered by factors such as statin therapy, viral infections, or cancer [38, 39, 42]. Although the precise mechanisms remain unclear, specific genetic associations, such as the HLA-DRB1*11:01 allele in adult patients with anti-HMGCR autoantibodies, have been identified. ILD and cardiac complications, particularly in individuals with anti-SRP antibody, have also been reported. Treatment focuses on addressing the underlying cause, such as statin withdrawal, or managing co-existing malignancies. There is good response to multiple-agent, long-term immunosuppressive therapies starting at high dose corticosteroids, as well as intravenous gamma globulins (IVIG) and rituximab, among others. It is said that IMNM is the most severe idiopathic inflammatory myopathy when it comes to muscle damage, and frequent relapses, such that chronicity could be expected [42].
3.1.2.3. Inclusion Body Myositis (IBM)
Inclusion body myositis (IBM) is a slowly progressive muscle disease primarily affecting older men patients aged 45 years and above. It presents with asymmetric muscle weakness, with distal muscles being more affected than the proximal ones. Commonly involved muscles include knee extensors and long finger flexors, and early muscle atrophy is observed. Facial weakness and dysphagia may occasionally occur. Muscle biopsy may reveal rimmed vacuoles, but additional markers such as TDP-43, p62 and valosin containing protein (VCP) positivity indicate ongoing degeneration. Lymphocytic infiltration around non-necrotic muscle fibers is typically present. IBM is considered to be an autoimmune and degenerative disease. The HLA locus is said to be the strongest risk alleles in the development of IBM. Its locus contains genes that encode MHC class I and II. The presence of endomysial infiltration with plasma cells and CD8+ T lymphocytes as well as MHC I expressing fibres indicate an autoimmune mechanism. A highly diagnostic biomarker for this myopathy is when antibodies that are targeting cN-1A or cytosolic 5’-nucleotidase 1A are detected in the blood [24, 33].
Also implicated is mitochondrial DNA variation characterized by mitochondrial pathology, including large mtDNA deletions and duplications, reduced mtDNA copy number, and somatic coding single nucleotide variants. These findings suggest an accelerated mitochondrial muscle aging process in sporadic IBM, possibly related to chronic inflammation [27]. A study found that TOMM40 or very long poly-T repeat allele of the mitochondrial protein translocase of outer mitochondrial membrane 40 intronic polymorphism had an IBM disease onset much later in life [43]. Unlike other immune-mediated myositis, IBM is considered a neurodegenerative disorder, one reason why steroid treatment will not help.
3.1.2.4. Antisynthetase Syndrome (ASS)
Antisynthetase syndrome (ASS) is a rare idiopathic inflammatory myopathy characterized by myositis, symmetrical arthritis and ILD along with serum autoantibodies to aminoacyl-transfer RNA synthetases (anti-ARS). It predominantly affects females, with highly variable features, including arthralgia, raynaud phenomenon, heliotropic rash, distal esophageal dysmotility and mechanic's hands. ASS is more frequent in females, with a female to male ratio approximately 7:3, and a mean age of 48±15 years at disease onset. It has a prevalence estimate of 1/25,000-33,000 worldwide, provided by the premise that about a quarter of all idiopathic inflammatory myopathy patients may have ASS [44]. Currently, it is thought that the pathogenesis starts with a genetic predisposition, then with the lung involvement triggered by environmental factors as tobacco exposure, airborne contaminants, and infections [45]. The non-specific tissue damage activates the innate immune system that leads to the release of immunogenic neo-antigens. There would be activation of the adaptive immune system with the production of anti-ARS antibodies, and the spread of the immune response to targeted tissues such as the muscle, joint, skin. Anti-synthetase antibodies (ARS) target cytoplasmic aminoacyl-tRNA synthetases, which are responsible for the ATP-dependent process of attaching a single amino acid to its corresponding tRNA, thus facilitating accurate protein synthesis. The antibodies include anti-Jo-1 (histidyl-tRNA synthetase), anti-PL-7 (threonyl), antiPL-12 (alanyl), anti-EJ (glycyl), anti-OJ (isoleucyl), anti-KS (asparaginyl), anti-Zo (phenylalanyl) and anti-Ha (tyrosyl) [44].
At the start of the disease, respiratory symptoms (shortness of breath, coughing, dysphagia) are found in 40-60% of patients [46], which may be acute or progressive. Some patients develop clinically overt myositis while others have hypomyopathic or even amyopathic forms. At onset, 20-70% of patients have muscle weakness of the proximal and axial muscles, and many have myalgia and muscle stiffness, similar to the milder presentations of other idiopathic inflammatory myopathies. ASS is a chronic disease which requires long-term treatment, and with a guarded prognosis [47].
3.1.2.5. Overlap Myositis (OM)
Overlap myositis (OM) is a type of myositis characterized by weakness in the arms and legs, elevated muscle enzymes (such as CK) and an association with other collagen disorders like sjogren syndrome, systemic sclerosis, or systemic lupus erythematosus (SLE). Therefore, treatment is more geared toward these collagen disorders. The histological characteristics of OM involve perifascicular necrosis and the binding of MHC class I and class II antibodies to the sarcolemma in specific areas of the skeletal muscle [26]. It is unclear if the myopathology of OM is specific to a particular disease or if it categorizes any of the major subsets of myositis. However, there have been cases where overlap syndrome showed features of antisynthetase syndrome, as evidenced by the presence of autoantibodies against multiple transfer-RNA components [48].
3.1.2.6. Treatment for Autoimmune Myositis
Clinical experience taught us that the mainstays of treatment in autoimmune myositis include corticosteroids (e.g., intravenous pulse methylprednisolone, oral prednisone in sequence, alongside bone and peptic ulcer protection), methotrexate and steroid-sparing immunosuppressive agents (e.g., azathioprine, mycophenolate, etc) [49]. In view of adverse events (including steroid-induced myopathy and “disfiguring” Cushing’s effect) in long term use needed, oral pednisolone may be taken together with azathioprine for at least three months, thenceforth, the former be slowly tapered for about three months prior to discontinuation. The steroid sparing drug shall be deemed to be the maintenance drug thereafter. There could be patients requiring steroids and in such a situation, it could be administered every other day together with the daily steroid-sparing agent. If the patient does not respond to these medications or experiences severe extramuscular symptoms, second-line agents such as cyclosporine and tacrolimus may be used [50].
In some settings, IVIG /subcutaneous gamma globulin, may also be applied. Cyclophosphamide, often in combination with other immunomodulatory agents like rituximab and IVIG, is reserved for severe cases or systemic organ involvement [50].
Various biological agents have been studied in myositis, including abatacept and janus kinase (JAK) inhibitors like tofacitinib, ruxolitinib, and baricitinib. Abatacept, which blocks the interaction between antigen-presenting cells and T-lymphocytes, has shown beneficial effects in refractory DM and polymyositis (PM). JAK inhibitors have also demonstrated efficacy in myositis patients, with tofacitinib showing positive results in disease activity and skin manifestations. Ongoing trials are evaluating the effectiveness of baricitinib [51]. Genomic variations can also influence the response to medications commonly used in the treatment of autoimmune myositis. For example, variations in the gene encoding the enzyme thiopurine methyltransferase (TPMT) can impact the metabolism of medications like azathioprine, potentially leading to differences in drug efficacy and toxicity. Genetic testing for TPMT variants can help guide individualized treatment decisions [50].
Purportedly a neurodegenerative condition too, IBM resists immunosuppressive therapy, though a number of regimens have been tried. Glucocorticoids, biologicals, and disease-modifying antirheumatic drugs (DMARDs) have not shown significant improvement. The use of IVIg in IBM remains a subject of debate, as controlled trials have shown mixed results. Sirolimus, an mTOR inhibitor, has shown promise in IBM treatment by decreasing T-effector cell proliferation, preserving T-regulatory cells, and inducing autophagy.
Exercise and physical therapy will be a complementary care. The red flag in the management of autoimmune myositis is the co-morbid malignancy, perhaps even as a paraneoplastic syndrome. Astute clinical assessment is required and screening for a malignancy from elsewhere (e.g. positron emission tomography-computerized scan [PET-CT scan]) may be key for early detection and management. In the absence of a malignancy elsewhere, a surveillance stance be the way for at least 5 years. Treatment for myositis should be tailored to the individual's disease activity and regularly monitored using appropriate scales, questionnaires, and muscle force testing. Moderate physical training is an important part of the treatment and has been shown to improve muscle weakness. In cases of extramuscular manifestations, interdisciplinary care involving neurologists, rheumatologists, dermatologists, pathologists, pulmonologists, and physical therapists is crucial for optimal patient management.
3.1.3. Metabolic Myopathies
Metabolic myopathies encompass a group of distinct disorders that impact various cellular energy metabolism due to deficiencies in a transport protein or enzymes [52]. The overall minimum incidence of metabolic diseases in children born in British Columbia is approximately 40 cases per 100,000 live births [53]. The surveyed data from thus study [53] is limited to specific metabolic disease groups. Around 24 children per 100,000 births (about 60% of the surveyed groups) have metabolic diseases involving amino acids, organic acids, primary lactic acidosis, galactosemia, or urea cycle disorders. Latest literature suggests the incidence of GSDs is approximately 1 case per 20000 to 43000 live births [40].
Cellular energy production relies on glycogen, glucose and free fatty acids, which play crucial roles in generating adenosine triphosphate (ATP). Pyruvate, a byproduct of glycogen metabolism, enters the mitochondria. While the carnitine transporter system facilitates the entry of long-chain fatty acids into the mitochondria, while short- and medium-chain fatty acids have unrestricted access. The carnitine transporter system involves acylcarnitine translocase, and carnitine palmitoyl transferases I and II. Once inside the mitochondria, these substrates undergo metabolism to produce acetyl coenzyme A, a vital component of ATP synthesis through the Krebs cycle. Any impairment in these biochemical pathways leads to reduced ATP levels within the muscles, resulting in weakness, muscle cramps particularly during exercise, myalgia, exercise intolerance and potentially rhabdomyolysis.
Numerous enzymes in the glycolytic and glycogenolytic pathway have then been associated with metabolic myopathies. Likewise, defects in fatty acid oxidation can affect free fatty acid transport or beta oxidation. While most patients primarily exhibit myopathic symptoms, some may present with predominant central nervous system manifestations, such as in glutaric aciduria type II. Mitochondriopathies refer to metabolic genetic defects that impact the electron transport chain, resulting in exercise intolerance, with or without rhabdomyolysis or fixed weakness. Diagnosing these disorders poses a challenge due to common clinical manifestations like exercise intolerance and myoglobinuria, necessitating the recognition of distinct features.
Exercise intolerance at the onset suggests a problem in glycogen metabolism, whereas a defect in fatty acid oxidation affects patients after prolonged exercise. Defects in glycogen metabolism can vary, with McArdle disease (glycogen storage type V), being the most common. It is an autosomal recessive disorder characterized by myophosphorylase deficiency. Individuals with this disease experience muscle cramps, fatigue, contractures and myoglobinuria. They are unable to tolerate static or isometric muscle contractions, as well as dynamic exercises. A pathognomonic sign of McArdle disease is the “second wind” phenomenon, where there is significant improvement in exercise tolerance about 10 minutes into exertional activities involving large muscle groups such as running. Exertional tachycardia decreases from a heart rate of 140 beats per minute to 120 beats per minute. This is due to the lack of glycogen stores in McArdle disease, leading the body to rely on the use of fatty acids as the main source of energy, which produces ATP at a slower rate, compared to glycogen. Symptom improvement can be achieved with sugar intake. Avoidance of activities that can trigger symptoms and a high carbohydrate diet is recommended [54].
Another important GSD is Pompe’s disease (glycogen storage type IIa), characterized by glycogen accumulation in the lysosome’s due alpha-glucosidase enzyme deficiency. This autosomal recessive disorder predominantly affects the skeletal and cardiac muscles. In the infantile form, symptoms include hypotonia, cardiomegaly and respiratory failure. The adult form is characterized by fixed muscle weakness and respiratory failure. Muscle biopsy reveals glycogen accumulation, correlating with the severity of clinical symptoms [55]. Enzyme replacement therapy with alglucosidase alfa (myozyme) improves outcomes, as it aids in the absorption and digestion of glycogen. In August 2021, avalglucosidase alfa was first approved in the United States for the treatment of late-onset Pompe disease [56].
Carnitine palmitoyltransferase II (CPT II) deficiency is the most common lipid storage disease [57]. Long-chain fatty acids require transport across the inner mitochondrial membrane mediated by CPT I and II, as well as acylcarnitine translocase, while short and medium chain fatty acids can freely cross into the mitochondria. This autosomal recessive disorder typically manifests symptoms from late childhood to adulthood. Patients experience frequent episodes of myalgias, muscle stiffness, weakness and myoglobinuria, often triggered by prolonged fasting or exercise and infection. Patients are asymptomatic between attacks, as fatty acids are the primary energy source for resting muscles and prolonged activities, patients manifest their symptoms at the end of rigorous exercise. Symptoms may also be provoked by stressors such as fasting, infections, fever and cold exposure. A high carbohydrate diet improves exercise endurance.
When evaluating metabolic myopathies, the serum total CK may be slightly elevated, especially in individuals at high risk of rhabdomyolysis. In phosphofructokinase deficiency, complete blood count and reticulocyte count may show signs of hemolytic anemia. A needle EMG may show myotonic discharges in acid maltase deficiency. Generally, needle EMG may reveal short-duration, low-amplitude motor unit action potentials. However, EMG readings may be normal in many metabolic myopathies.
3.1.4. Muscle Channelopathies
Skeletal muscle channel myopathies encompass rare and diverse conditions by mutations in the genes responsible for ion channels that enable proper functioning. These include SCN4A (sodium channels), CLCN1 (chloride channels), KCNJ2 and KCNJ18 (potassium channels), as well as CACNA1S (calcium channels) [58]. They can be classified into periodic paralyses (Andersen-Tawil syndrome, hyperkalemic periodic paralysis, and hypokalemic periodic paralysis) and the non-dystrophic myotonias (myotonia congenita, sodium channel myotonia, and paramyotonia congenita). Symptoms can emerge during the first and second decades or later in life, including during pregnancy [59]. These myopathies manifest as paroxysmal symptoms ranging from exaggerated muscle contraction to weakness due to disruptions in muscle membrane excitability. Overlapping clinical presentations make gene identification and treatment selection challenging.
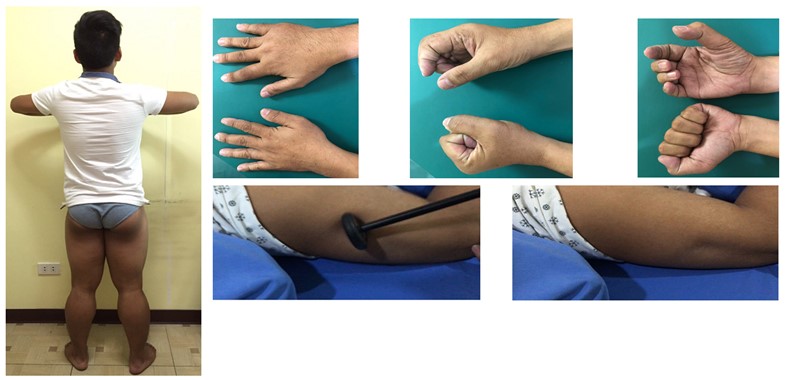
Note: Myotonia with delayed relaxation in grip (upper panel, sequential) and percussion (lower panel, sequential).
3.1.4.1. Nondystrophic Myotonias
Nondystrophic myotonias (NDMs) result from gain-of-function mutation in the SCN4A gene or loss-of-function mutation in the CLCN1 gene (illustrative case in Figure 2). Generally associated with grip, percussion, temperature aggravated and electrical myotonias (waxing-waning, “dive bomber sound” due to delayed muscle relaxation), these disorders encompass myotonia congenita (“herculean” muscles; autosomal dominant [Thomsens] and recessive [Becker] chloride channelopathies), sodium channel myotonias (includes congenital myopathy) and paramyotonia congenita (worsens with exercise and cold temperature; Eulenberg disease). Clinically, these are seen as muscle stiffness after voluntary contraction or percussion. With a prevalence of only <1:100,000 people, these disorders usually occur within the first two decades of life, and in the absence of progressive muscle wasting. These may be inherited either in an autosomal dominant or autosomal recessive manner. Currently, there are no FDA approved treatments, and that only certain symptomatic management strategies are available such as addressing triggers, as well as maintaining non-sedentary behavior. Symptomatic treatments to reduce muscle stiffness in NDM include carbamazepine, phenytoin, mexilitine, dantrolene, quinine, acetazolamide, trimeprazine and retigabine [60].
NDM should not be mistaken for another myotonic disorder in muscle (also with grip, percussion and electrical myotonias) accompanied by multi-organ disorders (baldness, cataracts, cardiac disease and insulin resistance, among others), called myotonic dystrophies (types 1 [distal dominant weakness plus facial and ocular muscle weakness] and 2 [proximal dominant weakness], with DMPK and CNBP mutations, respectively). The pseudomyotonias (grip myotonia but not with the typical percussion myotonia and electrical myotonia) are seen in a nerve terminal hyperexcitability disorder, Isaac’s disease/neuromyotonia. The latter is a VGKC channelopathy associated with myokymias hyporeflexia, hyper-sweating and disabling muscle stiffness. Immune therapies will be choice treatment for VGKC channelopathies, whether paraneoplastic and/or associated with dementia (Morvan syndrome).
3.1.4.2. Primary Periodic Paralyses
Primary periodic paralyses encompass disorders caused by mutations in sodium, potassium, and calcium channel gene mutations. Aberrant depolarization leads to inactivated sodium channels, resulting in reduced muscle membrane excitability and episodic generalized or focal weakness. These disorders, such as hyperkalemic periodic paralysis, hypokalemic periodic paralysis, and Anderson-Tawil syndrome, are autosomal dominant. Diagnosis may involve a history of flaccid paralysis and changes in serum potassium levels. Dichlorphenamide, and carbonic anhydrase inhibitors/acetazolamide have shown efficacy in treating primary periodic paralyses [58-60].
3.1.4.3. Secondary Periodic Paralysis
Secondary periodic paralysis occurs due to potassium wasting (related to factors like high carbohydrate meal, intense exercise, diuretic use, renal/adrenal disorders, licorice diet) and thyrotoxicosis. Cranial muscles are typically unaffected, and quadriparesis is often observed in the morning. Electrophysiologic short and prolonged exercise tests can aid in differentiating, secondary periodic paralysis and channelopathies. Appropriate treatment involves correcting electrolyte and hormone abnormalities by addressing the underlying disorders affecting key organs like the kidneys, adrenal glands or thyroid glands.
3.1.5. Drug-Induced Myopathies
Drug-induced myopathy refers to the acute or subacute onset of muscle weakness, myalgia, elevated CK levels, and myoglobinuria following the use of certain medications at therapeutic doses. Discontinuing the implicated medication typically resolves the myopathy. Numerous drugs can induce myopathy, including chloroquine, amiodarone, among others. These myotoxic substances may affect muscle organelles (e.g. myofibrillar proteins), may induce systemic effects (e.g. malabsorption or electrolyte imbalances) and may induce an inflammatory response [61].
Statin use is one of the most common causes of drug-induced myopathy. Statins are cholesterol-lowering drugs that inhibits hydroxymethylglutaryl-CoA (HMG-CoA) reductase. They are commonly prescribed for dyslipidemia, diabetes mellitus, stroke, hypertension and coronary artery disease. Skeletal muscle adverse effects are said to occur in 5-10% of patients [62]. Statin-associated muscle symptoms (SAMS) manifest as muscle tenderness, cramps, weakness and significantly elevated CK level (up to 10 times the upper limit of the normal), and in some cases rhabdomyolysis. Statins are known to affect the sarcoplasmic reticulum and mitochondria of type II muscle fibers. These fibers have less fat content compared to type I fibers, making them more susceptible to damage caused by reduced cholesterol availability for membrane production and biosynthesis. Statins also induce an immunologic reaction by altering the regulatory expression of T cells and B cells, leading to autoantibody production. The highest incidence of statin-induced myopathy is with simvastatin, while rosuvastatin and fluvastatin have the lowest. Symptoms are typically reversible upon discontinuation of statin therapy [62].
Colchicine intake in patients with gout, particularly those with renal insufficiency or concomitant use of nephrotoxic drugs like cyclosporine, can cause myopathy. Symptoms include proximal muscle weakness, elevated CK level, areflexia and sometimes sensory symptoms. Colchicine interferes with microtubule growth, and long-term use can lead to vacuolar myopathy with accumulation of autophagic vacuoles and lysosomes, as well as axonal neuropathy. Symptoms typically resolve within 4 to 6 weeks after discontinuing the medication.
Steroid-induced myopathic weakness is another adverse effect seen with chronic administration of prednisone or dexamethasone. Patients experience bilateral quadriceps weakness, and while the weakness is generally mild and may spare the neck flexor muscles, it can worsen pre-existing weaknesses especially in cancer patients. CK level and EMG findings are usually normal which is why it is not considered a true myopathy. On muscle biopsy there is only atrophy of type II fibers [31]. Lowering the dose may reverse the myopathic weakness. A clinical syndrome marked by myalgia, eosinophilia and scleroderma-like skin manifestations have been associated with L-tryptophan use. The syndrome mimics eosinophilic fasciomyositis (Shulman’s syndrome), the latter being responsive to immunosuppressive therapy.
There should be awareness too that cancer treatment with immune checkpoint inhibitors (e.g. pembrolizumab, nivolumab, etc.) may elicit immune-mediated myopathy (and even myasthenia), and muscle biopsies may show necrosis and mitochondrial abnormalities.
3.1.6. Intensive Care (ICU) Acquired Weakness
The clinical definition of intensive care unit-acquired weakness (ICUAW) is a noted weakness in critically ill patients and where the only etiologically plausible cause is the critical illness itself, and upon which weakness may persist long after discharge from ICU. Sepsis syndrome/or shock, multiple organ failure, metabolic variables like hyperglycemia, and duration of mechanical ventilation (MV) are among the risk factors for ICUAW. Attributable to weakness are impaired muscle contractility, muscle wasting, neuropathy, and pathways related to ubiquitin proteasome system and dysregulated autophagy which are associated with muscle protein degradation. Moreover, a preferential loss of myosin, is a distinct feature [63].
By nosology, ICUAW include critical illness polyneuropathy (CIP), critical illness myopathy (CIM), and an overlap syndrome, critical illness polyneuromyopathy (CIPNM). Versus CIP, the diagnostic criteria for CIM include: Presence of critical illness; Limb weakness or difficulty weaning from MV (non-neuromuscular causes excluded); compound muscle action potentials (CMAP) less than 80% lower limit of normal in at least two nerves without conduction block; CMAP duration increased on nerve or direct muscle stimulation; sensory nerve action potentials are greater than 80% of lower limb of normal; needle EMG shows myopathic potentials with early or normal recruitment in awake/collaborative patients; absence of abnormal response to repetitive nerve stimulation; and muscle biopsy with evidence of myopathy (e.g. myosin loss or muscle necrosis). Current management strategies include: achievement of euglycemia (intensive insulin therapy has been shown to have a protective effect); early mobilization (has positive functional outcomes); functional electrical stimulation (robust studies still desired); and nutritional intervention (promote muscle protein synthesis in relation to immobility) [64].
3.1.7. Rhabdomyolysis
Rhabdomyolysis is an urgent care acute myopathy with high morbidity and mortality, if not attended to. The causes include direct muscle trauma, status epilepticus, initiation/high dose use of medications (e.g. statins, anticonvulsants, anti-psychotic agents), toxins, infections (e.g. Weil's disease), muscle ischemia, metabolic/electrolyte disorders, exertion or prolonged bed rest, genetic disorders and temperature-induced states (e.g. neuroleptic malignant syndrome [NMS] and malignant hyperthermia [MH]). The triad of rhabdomyolysis constitute myalgia, weakness and myoglobinuria. An elevated total CK level is considered the sensitive test for muscle injury–induced rhabdomyolysis.
The foremost treatment aim in suspected rhabdomyolysis is care to avoid acute kidney injury. Fluid management is crucial in preventing prerenal azotemia, given the risk of muscle compartment fluid accumulation and the complication of hypovolemia. Thus, 1.5 L/h rate of aggressive hydration is suggested, while cognizant of cardiac overload. As an option, consider a 500 mL/h saline solution to be alternated every hour with 500 mL/h of 5% glucose solution with 50 mmol of sodium bicarbonate for each subsequent 2-3 L of solution. Also important in management is urine alkalization for renal protection against the nephrotoxicity from myoglobinuria and hyperuricosuria. Therefore, one should achieve a urinary output goal of 200 mL/h, urine pH >6.5, and plasma pH <7.5. The other management strategies include reversal of inappropriate arteriolar vasodilation through arteriolar restoration of contractility in injured muscles (i.e. mainly correction of acidosis and hyperkalemia). Likewise, mobilizing intramuscular edema will protect integrity of muscles and decompress muscle compartments [65]. In a center retrospective study of surgical anesthesia among cases with dystrophinopathy, they found no cases of rhabdomyolysis upon application of total intravenous anesthesia and no evidence for or against volatile anesthetic usage in this patient population. It is to be emphasized that no anesthetic agent is risk free. Non-triggering anesthetics, barbiturates, benzodiazepines, propofol, ketamine, and fasting have resulted to rhabdomyolysis in certain cases, including use of succinylcholine for muscle relaxation [66].
3.2 Inherited Myopathies
Certain inherited myopathies are currently undergoing various phase treatment trials focusing on genetic and inflammatory approaches. Among them, Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are the most common and extensively studied early- onset muscular dystrophies. DMD is a severe, and progressive neuromuscular disease caused by mutations in the dystrophin gene [67]. It follows an X-linked recessive inheritance pattern, making it rare in females, with an incidence of approximately 1 in 3500 to 5000 live male births [68]. BMD, on the other hand, is also caused by the DMD gene mutation but allows for residual expression of dystrophin protein, resulting in milder symptoms compared to DMD [69, 70].
Dystrophin plays a crucial role in maintain the structural integrity and contractility of myofibers, as well as regulating calcium levels, and protecting against oxidative stress. Loss of dystrophin leads to myocyte necrosis [69, 70]. Additionally, dystrophin contributes to the stability of the plasma membrane in myofibers. The primary manifestation of DMD is muscle weakness, predominantly affecting the proximal muscles of the lower extremities. As the disease progresses, weakness becomes more generalized. While developmental milestones may be achieved normally in the early years of life, there may be delays in attaining these milestones, as well as growth rate impairment and muscle tone abnormalities during infancy. Most patients typically experience difficulties in ambulation, running, climbing the stairs, and frequent falling during the first 2-3 years of life, although late onset manifestations may occur (in the third or fourth decade of life). Pseudohypertrophy of the calves and wasting of the thigh muscles are classic early features of DMD. Other manifestations include waddling gait, frequent fractures due to falls, joint contractures, pharyngeal muscle weakness leading to aspiration and hypernasality, sphincter muscle weakness causing urine and stool incontinence, and cardiac involvement resulting in heart failure.
Training exercises may be beneficial in DMD but again the evidence remains uncertain. This needs further research to know whether these training exercises promote functioning and improve health-related quality of life [70]. There is also no concrete evidence regarding the use of assistive devices but there are some studies in orthopedic assistive devices that can help the patient with DMD. However, genetic counselling be important as will respiratory and nutritional care. While several potential medications or drugs are now being studied, prolonged use of oral steroids (with bone protection) have been there in use, and alluded to, in regional expert consensus [71]. Gene therapy, as discussed herein under, involving the insertion of a dystrophin gene through a vector, has proven effective in animals but not humans. Ataluren, which is under a clinical study, is a molecule that binds with ribosomes and may allow the insertion of an amino acid in the premature termination codon, and producing a dystrophin that is smaller but functional [72].
The most common complications of this disease include those of decreased muscle function, spinal deformities, cardiovascular and respiratory compromise, and affected bone health. Therefore, it is important to note that a multidisciplinary approach must be undertaken to optimize the living conditions of patients suffering from this condition. Much importance must be given to identifying and targeting possible aspects that may be improved through either medical, technological, or engineering interventions [73]. Studies have shown that although DMD is a progressive disease, life expectancy rates have improved with advances in technology and medical treatment [74].
Therapeutic approaches for DMD aim to restore partially functional muscle dystrophin through three main strategies: gene delivery using viral vectors, stop codon read-through, and converting out-of-frame mutations to in-frame mutations (exon skipping) [75]. First is gene delivery using viral vectors: viral vectors, particularly adeno-associated virus (AAV), are used to deliver modified smaller versions of dystrophin called micro-dystrophins. These semi-functional proteins are about one-third the size of normal dystrophin. Preclinical studies in animal models have shown functional benefits, and early clinical trials have demonstrated high-level expression of micro-dystrophin in DMD patient muscles. Stop codon read-through: Approximately 10-15% of DMD cases have mutations that introduce premature stop codons, leading to truncated and non-functional dystrophin. Stop codon read-through drugs, such as ataluren, aim to enable the ribosome to insert an amino acid at the premature stop codon, potentially restoring dystrophin production. Clinical trials have shown variable improvements in walking ability, but drug-responsive increases in dystrophin have not been consistently demonstrated [76].
Exon skipping aims to convert out-of-frame mutations to in-frame mutations by blocking the inclusion of specific exons in the dystrophin pre-mRNA using oligonucleotide drugs. This process allows for the production of a partially functional dystrophin protein. Different oligonucleotide chemistries, such as 2'-O-methyl phosphorothioate (2OMePS) and phosphorodiamidate morpholino oligomer (PMO), have been studied. PMOs have shown higher potency in driving exon skipping and have been delivered systemically, resulting in notable dystrophin rescue in preclinical and clinical studies [76]. These are antisense oligonucleotides (ASO) that are approved for DMD patients given intravenously once a week.
Eteplirsen is an ASO designed for exon 51 skipping and was the first ASO to receive accelerated marketing authorization from the FDA for the treatment of DMD. The recommended dose of eteplirsen is 30 mg/kg administered once weekly as an intravenous (IV) infusion [68]. Golodirsen is used for exon 53 skipping. In a long-term safety and efficacy study of golodirsen in France, Italy and United Kingdom, a dose of 30 mg/kg/week, is well-tolerated in DMD patients. Adverse events (AEs) were generally mild, nonserious, and unrelated to golodirsen [77]. A phase 3, placebo-controlled, double-blind study (NCT02500381) is currently underway.
Viltolarsen is another ASO used for exon 53 skipping. The data were obtained from a long-term extension study and compared to a historical control group from the duchenne natural history study (DNHS), matched based on factors such as age, corticosteroid treatment, ambulatory ability, and geographic location [78]. The findings indicate that viltolarsen's effect on motor function was maintained over the 2-year period and support its safety profile, which was consistent with the observations from the 24-week phase 2 trial and suggests that viltolarsen maintains clinically relevant motor function over a 2-year period and has a similar safety profile. It highlights the emerging long-term data on FDA-approved DMD therapies and their effects on functional outcomes.
Casimersen is an ASO developed for exon 45 skipping. In a phase 1/2 study, casimersen, a drug used for exon 45 skipping in patients with duchenne muscular dystrophy (DMD), was found to be well tolerated. The reported treatment-emergent adverse events (TEAEs) were minimal and mostly mild, nonserious, and unrelated to casimersen. The observed adverse events were consistent with conditions commonly seen in children or as complications of DMD itself. No specific safety concerns related to casimersen, such as leukopenia, neutropenia, hepatotoxicity, severe skin reactions, infusion-site reactions, renal events, or cardiac events, were identified [79].
Exon skipping drugs restores the reading frame to allow DMD patients to make BMD-like residual dystrophin protein. It offers the possibility to restore the levels of residual functional dystrophin and can slow disease progression however it cannot restore muscle that was already lost. Renal function should be monitored in these patients because of the nephrotoxic side effect ASO. Overall, these therapeutic approaches hold promise for treating DMD by either delivering modified dystrophin genes, promoting read-through of premature stop codons, or modifying RNA splicing through exon skipping. Further research and clinical trials are needed to optimize these strategies and determine their long-term efficacy and safety in DMD patients.
4. Summary Statement and Conclusion
Myopathies are a diverse group of muscle disorders that can be acquired or inherited, leading to muscle weakness, impaired mobility, and other associated symptoms. These conditions can be challenging to diagnose and manage due to their varied etiologies and clinical presentations. Genetic myopathies, such as DMD and BMD, result from mutations in specific genes and often present in childhood. These conditions are characterized by progressive muscle degeneration and are currently the focus of extensive research and therapeutic development. Emerging treatments, such as gene therapy and exon skipping techniques, offer promising avenues to slow disease progression and improve the quality of life for affected individuals.
Acquired myopathies can arise from various factors, including medication use, critical illness, immune mechanisms, or metabolic disturbances. Drug-induced myopathies, often associated with medications like statins or steroids, can be resolved by discontinuing the offending drugs. On the other hand, critical illness myopathies, such as those occurring in intensive care unit (ICU) settings, require comprehensive management addressing the underlying condition, optimizing metabolic parameters, and providing supportive care. In managing myopathies, a multidisciplinary approach is crucial. This approach enables comprehensive evaluation, accurate diagnosis, and tailored treatment plans for individuals with myopathies (Table 4). While significant progress has been made in understanding myopathies and developing treatment strategies, further research is needed to advance our knowledge and improve therapeutic options. Continued efforts in genetic research, drug development, and supportive care strategies can potentially lead to more effective treatments and better outcomes for individuals affected by myopathies.
TABLE 4: Summary of myopathies with recommended treatment
|
Myopathy |
Recommended treatment |
|
Viral myositis HTLV-1 myositis HIV-associated myositis COVID-19 myopathy |
Supportive care, antiviral therapy, vaccination
-Steroids, interferon, danazol, high-dose vitamin C, antivirals -Antiretroviral therapy, immune modulators |
|
Bacterial myositis |
Penicillins, cephalosporins, vancomycin, gentamicin, clindamycin,
surgical drainage of pus |
|
Parasitic myositis |
Praziquantel, albendazole, pyrimethamine, sulfadiazine |
|
Fungal myositis |
Surgical debridement, amphotericin B, triazoles, echinocandins,
flucytosine |
|
Autoimmune myositis Dermatomyositis Antisynthetase syndrome Immune mediated necrotizing myopathy Overlap myositis Inclusion body myositis |
Corticosteroids, immunosuppressive drugs, IVIG, physical therapy, Rituximab and newer targeted therapies (e.g. Abatacept, Janus kinase
inhibitors)
-Supportive care, physical therapy, IVIG trial
|
|
Metabolic Myopathy |
-Enzyme replacement therapy, avoidance of triggers |
|
Muscle channelopathies |
-Symptomatic treatments to reduce muscle stiffness in NDM include
Carbamazepine, Phenytoin, Mexilitine, Dantrolene, Quinine, Acetazolamide,
Trimeprazine and Retigabine |
|
Drug-induced myopathies |
-Discontinuation of drug, supportive care |
|
ICU acquired weakness |
-Intensive insulin treatment, supportive care and early rehabilitation |
|
Rhabdomyolysis |
-Fluid management, urine alkalization, correction of acidosis and
hyperkalemia, decompression of muscle compartments |
|
Duchenne muscular dystrophy Becker muscular dystrophy |
-Prolonged use of steroids, gene therapy, stop codon read-through, exon
skipping therapy (Eteplirsen, Golodirsen, Viltolarsen, Casimersen) |
In conclusion, the field of myopathies is evolving, especially with advancements in diagnosis and patient care. Clinicians are made aware that there are potentially treatable myopathies, in which care ranges from targeted infectious agent medications, immune modulation treatments and gene therapies, to symptomatic care strategies, whether pharmacologic or non-pharmacologic. Rehabilitation and multi-disciplinary care also figure in the over-all management of patients suffering from myopathies.
REFERENCES
[1] Nidhi Garg, Susanna B Park, Steve Vucic, et al.
“Differentiating lower motor neuron syndromes.” J Neurol Neurosurg
Psychiatry, vol. 88, no. 6, pp. 474-483, 2017. View at: Publisher Site | PubMed
[2] Divisha Raheja, Charles Specht, Zachary Simmons
“Paraproteinemic neuropathies.” Muscle Nerve, vol. 51, no. 1, pp. 1-13,
2015. View at: Publisher Site | PubMed
[3] Nanna Witting, Linda K Andersen, John Vissing “Axial
myopathy: an overlooked feature of muscle diseases.” Brain, vol. 139,
no. Pt 1, pp. 13-22, 2016. View at: Publisher Site | PubMed
[4] Rose M Domingo-Horne, Mohammad Kian Salajegheh “An
Approach to Myopathy for the Primary Care Clinician.” Am J Med, vol.
131, no. 3, pp. 237-243, 2018. View at: Publisher Site | PubMed
[5] Siamak Moghadam-Kia, Chester V Oddis, Rohit Aggarwal
“Approach to asymptomatic creatine kinase elevation.” Cleve Clin J Med,
vol. 83, no. 1, pp. 37-42, 2016. View at: Publisher Site | PubMed
[6] L G Rider, F W Miller “Laboratory evaluation of the
inflammatory myopathies.” Clin Diagn Lab Immunol, vol. 2, no. 1, pp.
1-9. View at: Publisher Site | PubMed
[7] Steven Lovitt, Sandra L Moore, Franklin A Marden
“The use of MRI in the evaluation of myopathy.” Clin Neurophysiol, vol.
117, no. 3, pp. 486-495, 2006. View at: Publisher Site | PubMed
[8] Jemima Albayda, Nens van Alfen “Diagnostic Value of
Muscle Ultrasound for Myopathies and Myositis.” Curr Rheumatol Rep, vol.
22, no. 11, pp. 82, 2020. View at: Publisher Site | PubMed
[9] Nancy F Crum-Cianflone “Bacterial, Fungal,
Parasitic, and Viral Myositis.” Clin Microbiol Rev, vol. 21, no. 3, pp.
473-494, 2008. View at: Publisher Site | PubMed
[10]
Madu N
Soares, Moritz Eggelbusch, Elie Naddaf, et al. “Skeletal Muscle Alterations in
Patients with Acute Covid-19 and Post-acute Sequelae of Covid-19.” J
Cachexia Sarcopenia Muscle, vol. 13, no. 1, pp. 11-22, 2022. View at: Publisher Site | PubMed
[11]
Gayathri
Narayanappa, Bevinahalli Nanjegowda Nandeesh “Infective Myositis.” Brain
Pathol, vol. 31, no. 3, pp. e12950, 2021. View at: Publisher Site | PubMed
[12]
P Agyeman,
A Duppenthaler, U Heininger, et al. “Influenza-associated myositis in children.”
Infection, vol. 32, no. 4, pp. 199-203, 2004. View at: Publisher Site | PubMed
[13]
Nathaniel P
Disser, Andrea J De Micheli, Martin M Schonk, et al. “Musculoskeletal
Consequences of COVID-19.” J Bone Joint Surg Am, vol. 102, no. 14, pp.
1197-1204, 2020. View at: Publisher Site | PubMed
[14]
Chaolin
Huang, Lixue Huang, Yeming Wang, et al. “6-month Consequences of COVID-19 in
Patients Discharged from hospital: A Cohort Study.” Lancet, vol. 397,
no. 10270, pp. 220-232, 2021. View at: Publisher Site | PubMed
[15]
Ahmad Saud,
R Naveen, Rohit Aggarwal, et al. “COVID-19 and Myositis: What We Know So Far.” Curr
Rheumatol Rep, vol. 23, no. 8, pp. 63, 2021. View at: Publisher Site | PubMed
[16]
Kushagra
Gupta, Gauri Shankar Sharma, Ashok Kumar “COVID-19 vaccination-associated
anti-Jo-1 syndrome.” Reumatologia, vol. 59, no. 6, pp. 420-422, 2021.
View at: Publisher Site | PubMed
[17]
Mario
Chueire de Andrade-Junior, Isabel Chateaubriand Diniz de Salles, Christina May
Moran de Brito, et al. “Skeletal Muscle Wasting and Function Impairment in
Intensive Care Patients With Severe COVID-19.” Front Physiol, vol. 12,
pp. 640973, 2021. View at: Publisher Site | PubMed
[18]
I Higuchi,
K Hashimoto, E Matsuoka, et al. “The Main HTLV-1-harboring Cells in the Muscles
of Viral Carriers with Polymyositis are not Macrophages but CD4 Lymphocytes.” Acta
Neuropathol, vol. 92, no. 4, pp. 358-361, 1996. View at: Publisher Site | PubMed
[19]
Samar N
El-Beshbishi, Nairmen N Ahmed, Samar H Mostafa,, et al. “Parasitic Infections
and Myositis.” Parasitol Res, vol. 110, no. 1, pp. 1-18, 2012. View at: Publisher Site | PubMed
[20]
Hector H
Garcia “Neurocysticercosis.” Neurol Clin, vol. 36, no. 4, pp. 851-864,
2018. View at: Publisher Site | PubMed
[21]
Oscar H Del
Brutto, Bettsy Y Recalde, Robertino M Mera “Incidence of Adult-Onset Epilepsy
and the Contributory Role of Neurocysticercosis in a Five-Year,
Population-Based, Prospective Study in Rural Ecuador.” Am J Trop Med Hyg,
vol. 106, no. 1, pp. 208-214, 2021. View at: Publisher Site | PubMed
[22]
S
Prieto-González, J M Grau “Diagnosis and Classification of Granulomatous
Myositis.” Autoimmun Rev, vol. 13, no. 4-5, pp. 372-374, 2014. View at: Publisher Site | PubMed
[23]
A Mathur, J
M Kremer “Immunopathology, Musculoskeletal Features and Treatment of
Sarcoidosis.” Curr Opin Rheumatol, vol. 5, no. 1, pp. 90-94, 1993. View
at: Publisher Site | PubMed
[24]
Jantima
Tanboon, Ichizo Nishino “Classification of Idiopathic Inflammatory Myopathies:
Pathology Perspectives.” Curr Opin Neurol, vol. 32, no. 5, pp. 704-714,
2019. View at: Publisher Site | PubMed
[25]
Rachel
Zeng, Stefanie Glaubitz, Jens Schmidt “Inflammatory myopathies: shedding light
on promising agents and combination therapies in clinical trials.” Expert
Opin Investig Drugs, vol. 30, no. 11, pp. 1125-1140, 2021. View at: Publisher Site | PubMed
[26]
Jens
Schmidt “Current Classification and Management of Inflammatory Myopathies.” J
Neuromuscul Dis, vol. 5, no. 2, pp. 109-129, 2018. View at: Publisher Site | PubMed
[27]
Janine A
Lamb “The Genetics of Autoimmune Myositis.” Front Immunol, vol. 13, pp.
886290, 2022. View at: Publisher Site | PubMed
[28]
Kun Huang,
Rohit Aggarwal “Antisynthetase syndrome: A distinct disease spectrum.” J
Scleroderma Relat Disord, vol. 5, no. 3, pp. 178-191, 2020. View at: Publisher Site | PubMed
[29]
Ivan
Bogdanov, Jana Kazandjieva, Razvigor Darlenski, et al. “Dermatomyositis:
Current concepts.” Clin Dermatol, vol. 36, no. 4, pp. 450-458, 2018.
View at: Publisher Site | PubMed
[30]
Frederick W
Miller, Robert G Cooper, Jiří Vencovský, et al. “Myositis Genetics
Consortium. Genome-wide association study of dermatomyositis reveals genetic
overlap with other autoimmune disorders.” Arthritis Rheum, vol. 65, no.
12, pp. 3239-3247, 2013. View at: Publisher Site | PubMed
[31]
Marinos C
Dalakas, Reinhard Hohlfeld “Polymyositis and dermatomyositis.” Lancet,
vol. 362, no. 9388, pp. 971-982, 2003. PMID: 14511932. View at: Publisher Site | PubMed
[32]
Madeline E
DeWane, Reid Waldman, Jun Lu “Dermatomyositis Part I: Clinical Features and
Pathogenesis.” J Am Acad Dermatol, vol. 82, no. 2, pp. 267-281, 2020.
View at: Publisher Site | PubMed
[33]
Albert
Selva-O'Callaghan, Iago Pinal-Fernandez, Ernesto Trallero-Araguás, et al.
“Classification and Management of Adult Inflammatory Myopathies.” Lancet
Neurol, vol. 17, no. 9, pp. 816-828, 2018. View at: Publisher Site | PubMed
[34]
Akinori
Uruha, Hans-Hilmar Goebel, Werner Stenzel “Updates on the Immunopathology in
Idiopathic Inflammatory Myopathies.” Curr Rheumatol Rep, vol. 23, no. 7,
pp. 56, 2021. View at: Publisher Site | PubMed
[35]
Akinori
Uruha, Atsuko Nishikawa, Rie S Tsuburaya, et al. “Sarcoplasmic MxA expression:
A valuable marker of dermatomyositis.” Neurology, vol. 88, no. 5, pp.
493-500, 2017. View at: Publisher Site | PubMed
[36]
Marinos C
Dalakas “Inflammatory Muscle Diseases.” N Engl J Med, vol. 372, no. 18,
pp. 1734-1747, 2015. View at: Publisher Site | PubMed
[37]
Jessica A
Day, Vidya Limaye “Immune-mediated necrotising myopathy: A critical review of
current concepts.” Semin Arthritis Rheum, vol. 49, no. 3, pp. 420-429,
2019. View at: Publisher Site | PubMed
[38]
Martin
Klein, Heřman Mann 1, Lenka Pleštilová,
et al. “Increasing Incidence of Immune-mediated Necrotizing Myopathy:
Single-centre Experience.” Rheumatology (Oxford), vol. 54, no. 11, pp.
2010-2014, 2015. View at: Publisher Site | PubMed
[39]
Iago
Pinal-Fernandez, Maria Casal-Dominguez, Andrew L Mammen “Immune-mediated
Necrotizing Myopathy.” Curr Rheumatol Rep, vol. 20, no. 4, pp. 21, 2018.
View at: Publisher Site | PubMed
[40]
William L.
Stone, Hajira Basit, Abdullah Adil “Glycogen Storage Disease.” In: StatPearls
[Internet]. Treasure Island (FL), StatPearls Publishing, 2023. View at: PubMed
[41]
Chen-Hua
Wang, Wen-Chen Liang “Pediatric immune-mediated necrotizing myopathy.” Front
Neurol, vol. 14, pp. 1123380, 2023. View at: Publisher Site | PubMed
[42]
Yves
Allenbach, Olivier Benveniste, Werner Stenzel, et al. “Immune-mediated
Necrotizing Myopathy: Clinical Features and Pathogenesis.” Nat Rev Rheumatol,
vol. 16, no. 12, pp. 689-701, 2020. View at: Publisher Site | PubMed
[43]
Qiang Gang,
Conceicao Bettencourt, Pedro M Machado, et al. “The Effects of an Intronic
Polymorphism in TOMM40 and APOE Genotypes in Sporadic Inclusion Body Myositis. Neurobiol
Aging, vol. 36, no, 4, pp. 1766.e1, 2015. View at: Publisher Site | PubMed
[44]
Aleksandra
Halina Opinc, Joanna Samanta Makowska “Antisynthetase Syndrome - Much More Than
Just a Myopathy.” Semin Arthritis Rheum, vol. 51, no. 1, pp. 72-83,
2021. View at: Publisher Site | PubMed
[45]
Nasam
Alfraji, Usman Mazahir 2, Moiuz Chaudhri, et al. “Anti-synthetase Syndrome: A
Rare and Challenging Diagnosis for Bilateral Ground-glass Opacities—A Case Report
with Literature Review.” BMC Pulm Med, vol. 21, no. 1, pp. 11, 2021.
View at: Publisher Site | PubMed
[46]
Siamak
Moghadam-Kia, Chester V Oddis “Current and New Targets for Treating Myositis.” Curr
Opin Pharmacol, vol. 65, pp. 102257, 2022. View at: Publisher Site | PubMed
[47]
Leah J
Witt, James J Curran, Mary E Strek “The Diagnosis and Treatment of
Antisynthetase Syndrome.” Clin Pulm Med, vol. 23, no. 5, pp. 218-226,
2016. View at: Publisher Site | PubMed
[48]
Haruhiko
Motegi, Yohei Kirino, Ryoji Morishita, et al. “Overlap syndrome with antibodies
against multiple transfer-RNA components presenting antisynthetase syndrome.” Neuromuscul
Disord, vol. 33, no. 5, pp. 405-409, 2023. View at: Publisher Site | PubMed
[49]
Hirokazu
Sasaki, Hitoshi Kohsaka “Current diagnosis and treatment of polymyositis and
dermatomyositis.” Mod Rheumatol, vol. 28, no. 6, pp. 913-921, 2018. View
at: Publisher Site | PubMed
[50]
Rachel
Zeng, Stefanie Glaubitz, Jens Schmidt “Antibody Therapies in Autoimmune
Inflammatory Myopathies: Promising Treatment Options.” Neurotherapeutics,
vol. 19, no. 3, pp. 911-921, 2022. View at: Publisher Site | PubMed
[51]
Iain B
McInnes, Nicole L Byers, Richard E Higgs, et al. “Comparison of baricitinib,
upadacitinib, and tofacitinib mediated regulation of cytokine signaling in
human leukocyte subpopulations.” Arthritis Res Ther, vol. 21, no. 1, pp.
183, 2019. View at: Publisher Site | PubMed
[52]
B T Darras,
N R Friedman “Metabolic myopathies: a clinical approach; part I.” Pediatr
Neurol, vol. 22, no. 2, pp. 87-97, 2000. View at: Publisher Site | PubMed
[53]
D A
Applegarth, J R Toone, R B Lowry “Incidence of inborn errors of metabolism in
British Columbia, 1969-1996.” Pediatrics, vol. 105, no. 1, pp. e10,
2000. View at: Publisher Site | PubMed
[54]
Zoia E.
Khattak, Muddasir Ashraf “McArdle Disease.” n: StatPearls [Internet]. Treasure
Island (FL): StatPearls Publishing, 2023. View at: PubMed
[55]
Lara
Kohler, Rosa Puertollano, Nina Raben “Pompe Disease: From Basic Science to
Therapy.” Neurotherapeutics, vol. 15, no. 4, pp. 928-942, 2018. View at:
Publisher Site | PubMed
[56]
Sohita
Dhillon “Avalglucosidase alfa: First Approval.” Drugs, vol. 81, no. 15,
pp. 1803-1809, 2021. View at: Publisher Site | PubMed
[57]
Pushpa Raj
Joshi, Stephan Zierz “Muscle Carnitine Palmitoyltransferase II (CPT II)
Deficiency: A Conceptual Approach.” Molecules, vol. 25, no. 8, pp. 1784,
2020. View at: Publisher Site | PubMed
[58]
Lauren
Phillips, Jaya R Trivedi “Skeletal Muscle Channelopathies.” Neurotherapeutics,
vol. 15, no. 4, pp. 954-965, 2018. View at: Publisher Site | PubMed
[59]
Emma
Matthews, Sarah Holmes, Doreen Fialho “Skeletal Muscle Channelopathies: A Guide
to Diagnosis and Management.” Pract Neurol, vol. 21,no. 3, pp. 196-204,
2021. View at: Publisher Site | PubMed
[60]
Bas C
Stunnenberg, Samantha LoRusso, W David Arnold, et al. “Guidelines on Clinical
Presentation and Management of Nondystrophic Myotonias.” Muscle Nerve,
vol. 62, no. 4, pp. 430-444, 2020. View at: Publisher Site | PubMed
[61]
M C Dalakas
“Toxic and Drug Induced Myopathies.” J Neurol Neurosurg Psychiatry, vol.
80, no. 8, pp. 832-838, 2009. View at: Publisher Site | PubMed
[62]
Silvia
Attardo, Olimpia Musumeci, Daniele Velardo, et al. “Statins Neuromuscular
Adverse Effects.” Int J Mol Sci, vol. 23, no. 15, pp. 8364, 2022. View
at: Publisher Site | PubMed
[63]
Heta Lad,
Tyler M Saumur, Margaret S Herridge, et al. “Intensive Care Unit-Acquired
Weakness: Not Just Another Muscle Atrophying Condition.” Int J Mol Sci,
vol. 21, no. 21, pp. 7840, 2020. View at: Publisher Site | PubMed
[64]
Kevin
Cheung, Alasdair Rathbone, Michel Melanson, et al. “Pathophysiology and
Management of Critical Illness Polyneuropathy and Myopathy.” J Appl Physiol
(1985), vol. 130, no. 5, pp. 1479-1489, 2021. View at: Publisher Site | PubMed
[65]
Patrick A
Torres, John A Helmstetter, Adam M Kaye, et al. “Rhabdomyolysis: Pathogenesis,
Diagnosis, and Treatment.” Ochsner J, vol. 15, no. 1, pp. 58-69, 2015.
View at: PubMed
[66]
Leal G
Segura, Jessica D Lorenz, Toby N Weingarten, et al. “Anesthesia and Duchenne or
Becker Muscular Dystrophy: Review of 117 Anesthetic Exposures.” Paediatr
Anaesth, vol. 23, no. 9, pp. 855-864, 2013. View at: Publisher Site | PubMed
[67]
Dongsheng
Duan, Nathalie Goemans, Shin'ichi Takeda, et al. “Duchenne Muscular Dystrophy.”
Nat Rev Dis Primers, vol. 7, no. 1, pp. 13, 2021. View at: Publisher Site | PubMed
[68]
Gökçe Eser,
Haluk Topaloğlu “Current Outline of Exon
Skipping Trials in Duchenne Muscular Dystrophy.” Genes
(Basel), vol. 13, no. 7, pp. 1241, 2022. View at: Publisher Site | PubMed
[69]
Salvatore
Crisafulli, Janet Sultana, Andrea Fontana, et al. “Global Epidemiology of
Duchenne Muscular Dystrophy: An Updated Systematic Review and Meta-analysis.” Orphanet
J Rare Dis, vol. 15, no. 1, pp. 141, 2020. View at: Publisher Site | PubMed
[70]
Stian
Hammer, Michel Toussaint, Maria Vollsæter, et al. Exercise Training in Duchenne
Muscular Dystrophy: A Systematic Review and Meta-Analysis. J Rehabil Med,
vol. 54, pp. jrm00250, 2022. View at: Publisher Site | PubMed
[71]
Fumi Takeuchi, Harumasa Nakamura, Naohiro Yonemoto, et al. “Clinical
Practice with Steroid Therapy for Duchenne Muscular Dystrophy: An Expert Survey
in Asia and Oceania.” Brain Dev,
vol. 42, no. 3, pp. 277-288, 2020. View at: Publisher Site | PubMed
[72]
Maria de
los Angeles Beytía, Julia Vry, Janbernd Kirschner “Drug Treatment of Duchenne
Muscular Dystrophy: Available Evidence and Perspectives.” Acta Myol,
vol. 31, no. 1, pp. 4-8, 2012. View at: PubMed
[73]
K. Bushby,
J. Bourke, R. Bullock, et al. “The Multidisciplinary Management of Duchenne
Muscular Dystrophy.” Curr Paediatr, vol. 15, no. 4, pp. 292-300, 2005.
View at: Publisher Site
[74]
Jonathan
Broomfield, Micki Hill, Michela Guglieri, et al. “Life Expectancy in Duchenne
Muscular Dystrophy: Reproduced Individual Patient Data Meta-analysis.” Neurol,
vol. 97, no. 23, pp. e2304-e2314, 2021. View at: Publisher Site | PubMed
[75]
Shin'ichi
Takeda, Paula R Clemens, Eric P Hoffman “Exon-Skipping in Duchenne Muscular
Dystrophy.” J Neuromuscul Dis, vol. 8, no. s2, pp. S343-S358, 2021. View
at: Publisher Site | PubMed
[76]
Omar
Sheikh, Toshifumi Yokota “Developing DMD therapeutics: a review of the
effectiveness of small molecules, stop-codon readthrough, dystrophin gene
replacement, and exon-skipping therapies.” Expert Opin Investig Drugs,
vol. 30, no. 2, pp. 167-176, 2021. View at: Publisher Site | PubMed
[77]
Laurent
Servais, Eugenio Mercuri, Volker Straub, et al. “Long-Term Safety and Efficacy
Data of Golodirsen in Ambulatory Patients with Duchenne Muscular Dystrophy
Amenable to Exon 53 Skipping: A First-in-human, Multicenter, Two-Part,
Open-Label, Phase 1/2 Trial.” Nucleic Acid Ther, vol. 32, no. 1, pp.
29-39, 2022. View at: Publisher Site | PubMed
[78]
Paula R
Clemens, Vamshi K Rao, Anne M Connolly, et al. “Long-Term Functional Efficacy
and Safety of Viltolarsen in Patients with Duchenne Muscular Dystrophy.” J
Neuromuscul Dis, vol. 9, no. 4, pp. 493-501, 2022. View at: Publisher Site | PubMed
[79]
Kathryn R
Wagner, Nancy L Kuntz, Erica Koenig, et al. “Safety, tolerability, and
pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy
amenable to exon 45 skipping: A randomized, double-blind, placebo-controlled,
dose-titration trial.” Muscle Nerve, vol. 64, no. 3, pp. 285-292, 2021.
View at: Publisher Site | PubMed
[80]
Kelly
Graham Gwathmey, Kathleen T Pearson “Diagnosis and management of sensory
polyneuropathy.” BMJ, vol. 365, pp. l1108, 2019. View at: Publisher Site | PubMed
[81]
Vitória
Pimentel, Vanessa Wallau Luchsinger, Gabriel Leal Carvalho, et al.
“Guillain-Barré syndrome associated with COVID-19: A systematic review.” Brain
Behav Immun Health, vol. 28, pp. 100578, 2023. View at: Publisher Site | PubMed
[82]
Srikanth
Muppidi, Steven Vernino “Paraneoplastic neuropathies.” Continuum (Minneap
Minn), vol. 20, no. 5 Peripheral Nervous System Disorders, pp. 1359-1372,
2014. View at: Publisher Site | PubMed
[83]
Laura A
Foster, Mohammad Kian Salajegheh. “Motor Neuron Disease: Pathophysiology,
Diagnosis, and Management.” Am J Med, vol. 132, no. 1, pp. 32-37, 2019.
View at: Publisher Site | PubMed
[84]
Mark A
Tarnopolsky “Metabolic Myopathies.” Continuum (Minneap Minn), vol. 28,
no. 6, pp. 1752-1777, 2022. View at: Publisher Site | PubMed
[85]
Eva L
Feldman, Brian C Callaghan, Rodica Pop-Busui, et al. “Diabetic neuropathy.” Nat
Rev Dis Primers, vol. 5, no. 1, pp. 42, 2019. View at: Publisher Site | PubMed
[86]
Jinny Tavee
“Peripheral neuropathy in sarcoidosis.” J Neuroimmunol, vol. 368, pp.
577864, 2022. View at: Publisher Site | PubMed
[87]
Rami Massie,
Michelle L Mauermann, Nathan P Staff, et al. “Diabetic cervical radiculoplexus
neuropathy: a distinct syndrome expanding the spectrum of diabetic
radiculoplexus neuropathies.” Brain, vol. 135, no. Pt 10, pp. 3074-3088,
2012. View at: Publisher Site | PubMed
[88]
Ryan
Newberger, Vikas Gupta Streptococcus Group A. In: StatPearls [Internet].
Treasure Island (FL): StatPearls Publishing, 2023. View at: PubMed
[89]
Sreenath
Meegada, Hamza Akbar, Suman Siddamreddy, et al. “Granulomatous Myositis
Associated with Extremely Elevated Anti-striated Muscle Antibodies in the
Absence of Myasthenia Gravis.” Cureus, vol. 12, no. 2, pp. e6981, 2020.
View at: Publisher Site | PubMed
[90]
Jon Andoni
Urtizberea, Gianmarco Severa, Edoardo Malfatti “Metabolic Myopathies in the Era
of Next-Generation Sequencing.” Genes, vol. 14, no. 5, pp. 954, 2023.
View at: Publisher Site | PubMed
[91]
Nuria
Carrillo, May C Malicdan, Marjan Huizing, et al. “GNE Myopathy: Etiology,
Diagnosis and Therapeutic challenges.” Neurotherapeutics, vol. 15, no.
4, pp. 900-914, 2018. View at: Publisher Site | PubMed
[92]
Yongpeng
Ge, Xin Lu, Qinglin Peng, et al. “Clinical Characteristics of
Anti-3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Antibodies in Chinese
Patients with Idiopathic Inflammatory Myopathies.” PLoS One, vol. 10,
no. 10, pp. e0141616, 2015. View at: Publisher Site | PubMed
[93] Jean-Luc Senécal, Jean-Pierre Raynauld, Yves Troyanov “Editorial: A New Classification of Adult Autoimmune Myositis.” Arthritis Rheumatol, vol. 69, no. 5, pp. 878-884, 2017. View at: Publisher Site | PubMed
[94] Renske G Kamperman, Anneke J van der Kooi, Marianne de Visser, et al. “Pathophysiological Mechanisms and Treatment of Dermatomyositis and Immune Mediated Necrotizing Myopathies: A Focused Review.” Int J Mol Sci vol. 23, no. 8, pp. 4301, 2022. View at: Publisher Site | PubMed